Table of Contents
In neonates, vomiting may range from benign physiological regurgitation to a surgical emergency. A systematic approach is essential.
1. Gastrointestinal Causes
A. Physiological / Functional
- Physiological gastroesophageal reflux (GER)
- Overfeeding
- Improper feeding technique
- Aerophagia (swallowed air)
- Delayed gastric emptying in preterm infants
B. Gastrointestinal Obstruction
High Intestinal Obstruction
Bilious vomiting is a surgical emergency until proven otherwise.
Esophageal
- Esophageal atresia ± tracheoesophageal fistula
- Esophageal stricture
- Congenital esophageal stenosis
Gastric
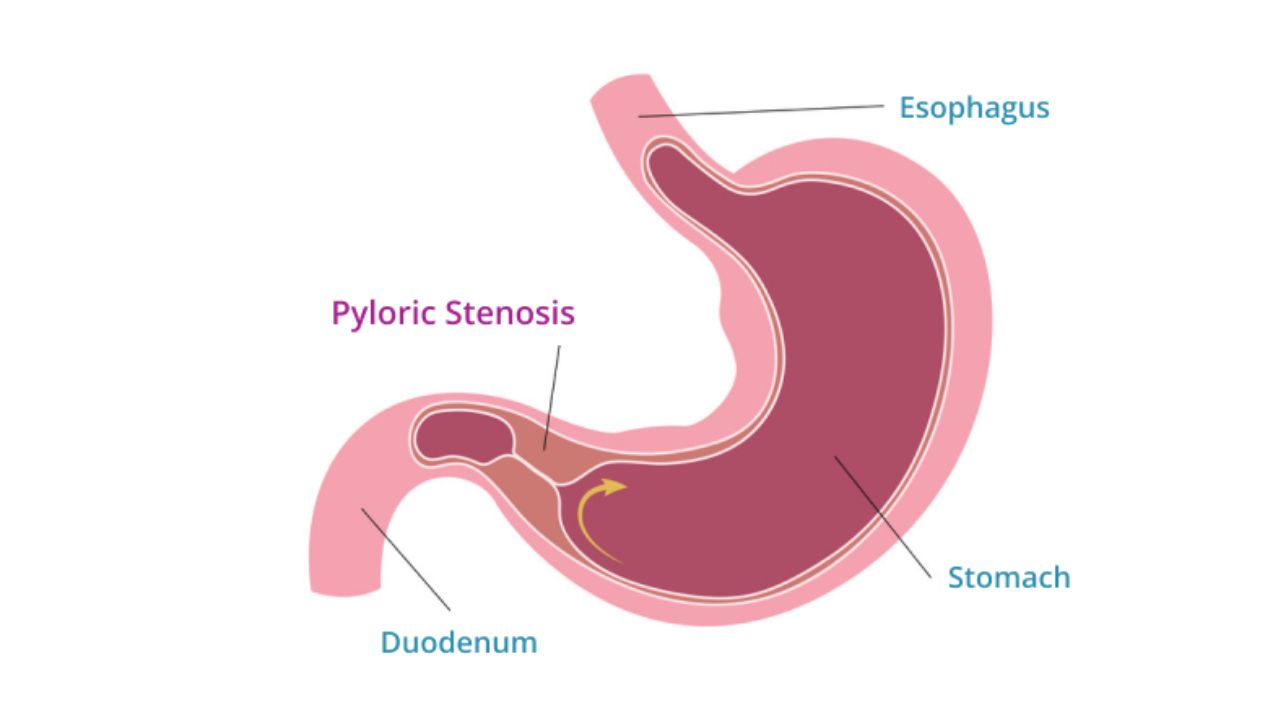
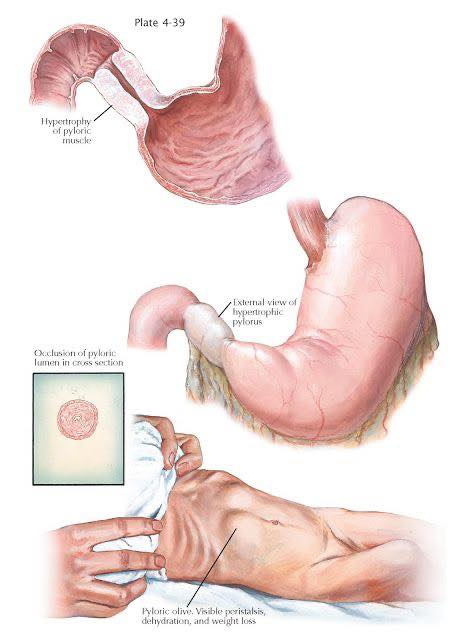
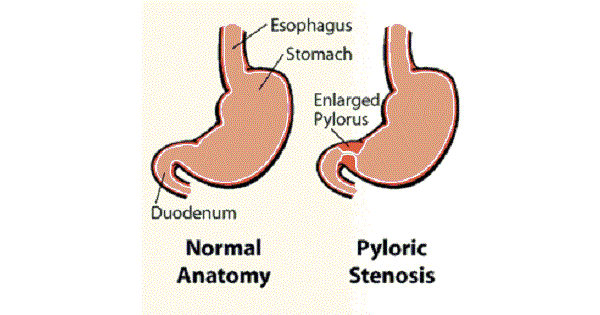
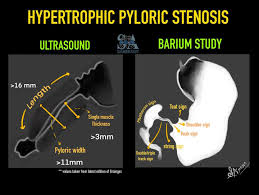
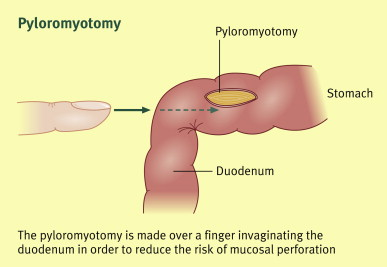
- Pyloric stenosis (typically 2–8 weeks)
- Gastric volvulus
- Gastric outlet obstruction
- Antral web
Duodenal
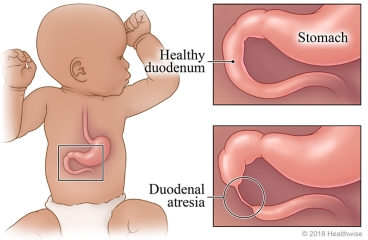
- Duodenal atresia
- Duodenal stenosis
- Annular pancreas
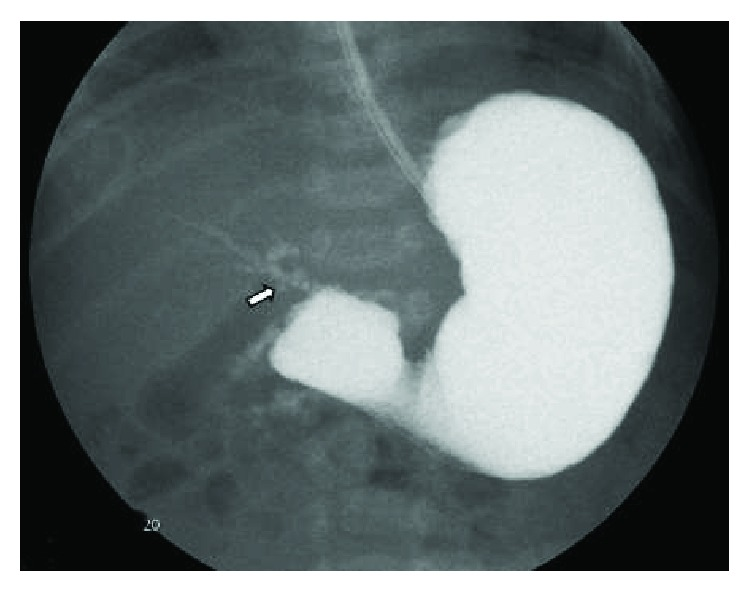
- Malrotation with midgut volvulus
- Ladd bands
Jejunal/Ileal
- Jejunal atresia
- Ileal atresia
- Meconium ileus
- Meconium plug syndrome
- Small left colon syndrome
Colonic
- Hirschsprung disease
- Colonic atresia
- Anorectal malformations
C. Inflammatory/Infectious GI Disease
Necrotizing Enterocolitis (NEC)
Common NICU cause:
- Vomiting
- Feed intolerance
- Abdominal distension
- Bloody stools
Spontaneous Intestinal Perforation
Enterocolitis
- Bacterial
- Viral
- Fungal
2. Infectious Causes
Any neonatal sepsis can present with vomiting.
Systemic Sepsis
- Early-onset sepsis
- Late-onset sepsis
Common organisms:
- Group B Streptococcus
- Escherichia coli
- Listeria monocytogenes
- Klebsiella
- Enterobacter
- Staphylococcus aureus
- CoNS
- Candida
CNS Infections
- Meningitis
- Encephalitis
- Brain abscess (rare)
Urinary Tract Infection
A very important cause of unexplained vomiting.
3. Metabolic and Endocrine Causes
Inborn Errors of Metabolism (IEM)
Consider especially when vomiting is associated with:
- Lethargy
- Acidosis
- Hyperammonemia
- Hypoglycemia
Disorders
Amino Acid Disorders
- Maple syrup urine disease
- Phenylketonuria
- Homocystinuria
Organic Acidemias
- Propionic acidemia
- Methylmalonic acidemia
- Isovaleric acidemia
Urea Cycle Disorders
- OTC deficiency
- CPS deficiency
Fatty Acid Oxidation Disorders
- MCAD deficiency
- VLCAD deficiency
Carbohydrate Disorders
- Galactosemia
- Hereditary fructose intolerance
Electrolyte Disorders
- Hyponatremia
- Hypernatremia
- Hypokalemia
- Hyperkalemia
- Hypocalcemia
- Hypercalcemia
- Hypomagnesemia
Glucose Disorders
- Hypoglycemia
- Hyperglycemia
Endocrine Disorders
Congenital Adrenal Hyperplasia (salt-wasting)
- Vomiting
- Dehydration
- Shock
Adrenal insufficiency
Congenital hypothyroidism
Hyperthyroidism (rare)
4. Neurological Causes
Raised intracranial pressure can cause vomiting.
Intracranial Hemorrhage
- Germinal matrix hemorrhage
- Intraventricular hemorrhage
- Subdural hemorrhage
Hydrocephalus
- Congenital
- Post-hemorrhagic
Hypoxic-Ischemic Encephalopathy
CNS Malformations
- Dandy-Walker malformation
- Arnold-Chiari malformation
Seizures
May manifest as feed intolerance and vomiting.
5. Respiratory Causes
Severe respiratory distress
- Respiratory distress syndrome
- Pneumonia
- PPHN
- Congenital heart disease with heart failure
Mechanism:
- Increased swallowed air
- Gut hypoperfusion
6. Cardiac Causes
Congenital Heart Disease
Particularly:
- Duct-dependent lesions
- Heart failure states
Examples:
- Coarctation of aorta
- Hypoplastic left heart syndrome
- Interrupted aortic arch
Congestive Cardiac Failure
- Large VSD
- PDA
- Cardiomyopathy
7. Drug-Related Causes
Maternal Drug Exposure
- Opioid withdrawal
- SSRI exposure
NICU Medications
- Caffeine
- Theophylline
- Erythromycin
- Opioids
- Iron supplements
- Vitamin preparations
8. Feeding-Related Causes
Feeding Intolerance
Common in preterm infants
Features:
- Vomiting
- Increased gastric residuals
- Abdominal distension
Human Milk Fortifier Intolerance
Formula Intolerance
Cow’s Milk Protein Allergy
Can present with:
- Vomiting
- Blood in stool
- Poor weight gain
9. Hepatobiliary and Pancreatic Causes
- Neonatal hepatitis
- Cholestasis
- Biliary atresia
- Pancreatitis (rare)
- Choledochal cyst
10. Toxic Causes
- Medication overdose
- Hypervitaminosis
- Accidental toxin exposure
Important NICU “Cannot Miss” Diagnoses
Any neonate with vomiting should be assessed urgently for:
- Malrotation with midgut volvulus
- Necrotizing enterocolitis (NEC)
- Sepsis
- Meningitis
- Congenital adrenal hyperplasia
- Inborn errors of metabolism
- Intestinal atresia
- Hirschsprung disease
- Pyloric stenosis
- Intracranial hemorrhage
Practical NICU Approach
Bilious Vomiting
Think:
- Malrotation with volvulus
- Intestinal atresia
- Hirschsprung disease
- Meconium ileus
- NEC
→ Surgical consultation immediately.
Non-bilious Projectile Vomiting
Think:
- Pyloric stenosis
- GER
- Overfeeding
Vomiting + Abdominal Distension
Think:
- NEC
- Obstruction
- Sepsis
Vomiting + Shock
Think:
- Sepsis
- CAH
- Volvulus
- Metabolic disease
Vomiting + Lethargy/Seizures
Think:
- Meningitis
- IVH
- Hypoglycemia
- IEM
- Electrolyte disturbance
For NICU practice, the highest-yield etiologies are GER/overfeeding, feeding intolerance of prematurity, NEC, sepsis, malrotation-volvulus, intestinal obstruction, CAH, and inborn errors of metabolism. These account for most clinically significant neonatal vomiting presentations.