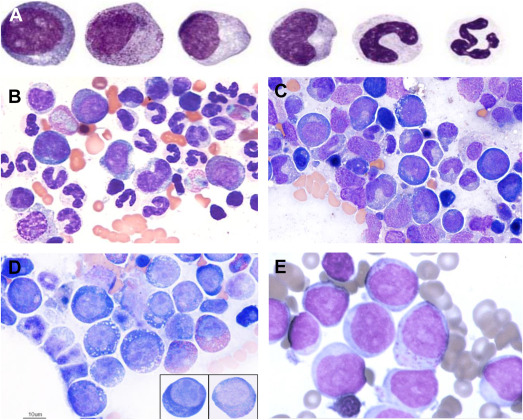
Diamond-Blackfan anemia (DBA) is a rare congenital blood disorder characterized by the failure of the bone marrow to produce red blood cells. It usually presents in infancy and is classified as a congenital pure red cell aplasia. DBA is notable for its genetic basis, variable physical malformations, and lifelong management challenges.
Key facts
Onset: Typically within the first year of life
Genetic cause: Mutations in ribosomal protein genes
Inheritance pattern: Autosomal dominant (most cases de novo)
Prevalence: About 5–7 per million live births
Treatment options: Corticosteroids, chronic transfusions, or stem cell transplantation
Pathophysiology
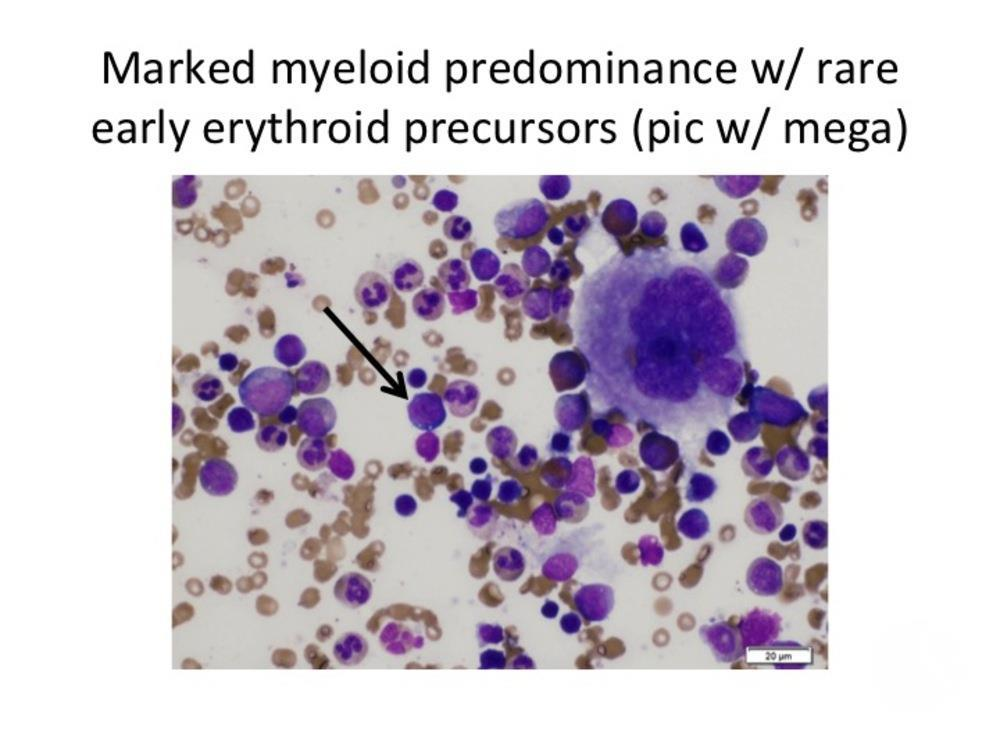
Diamond-Blackfan anemia arises from mutations that impair ribosome biogenesis, leading to defective erythroid progenitor development. The bone marrow becomes selectively deficient in red cell precursors, while white cells and platelets remain normal. Most cases involve mutations in genes encoding ribosomal proteins such as RPS19, RPL5, or RPL11, disrupting protein synthesis and cellular growth.
Clinical features
Infants with DBA commonly present with pallor and anemia. Physical anomalies are present in about half of cases, including craniofacial abnormalities, thumb or limb malformations, and heart or kidney defects. Growth retardation and an increased lifetime risk of malignancies such as leukemia and osteogenic sarcoma are recognized complications.
Diagnosis
Diagnosis combines hematologic findings—macrocytic anemia, reticulocytopenia, and normal marrow cellularity except for absent red cell precursors—with genetic testing for ribosomal protein gene mutations. Elevated erythrocyte adenosine deaminase (eADA) activity is a common biomarker.
Management and prognosis
Initial treatment often involves corticosteroids to stimulate red cell production. Patients unresponsive to steroids may require regular transfusions with iron chelation therapy to prevent overload, or hematopoietic stem cell transplantation as a potential cure. Advances in genetic understanding have improved prognosis, but lifelong monitoring remains essential due to treatment complications and cancer risk.
Vomiting is one of the most common presenting complaints in pediatric practice—ranging from benign, self-limiting illnesses to life-threatening surgical and metabolic emergencies. It is not a diagnosis, but a symptom with a broad differential, involving gastrointestinal (GI), neurological, metabolic, infectious, and psychological causes.
The real challenge is not treating vomiting—but identifying which child is sick and why.
Understanding Vomiting
Vomiting is a protective reflex involving coordinated contraction of abdominal muscles and relaxation of the lower esophageal sphincter, leading to expulsion of gastric contents.
Types of Vomiting
Acute vomiting → hours to days (e.g., gastroenteritis)
Bilious vomiting (green) → surgical emergency until proven otherwise
Step 1: Initial Stabilization (ABC First)
Before thinking of diagnosis:
Airway → risk of aspiration
Breathing → respiratory distress?
Circulation → shock, dehydration
Assess:
Vitals
Capillary refill
Level of consciousness
Hydration status
👉 This step is critical because vomiting can rapidly lead to dehydration and electrolyte imbalance.
Step 2: Identify RED FLAG Signs
These determine urgency and need for immediate intervention:
Major Red Flags
Bilious (green) vomiting
Bloody vomiting
Altered sensorium
Severe dehydration
Persistent projectile vomiting
Abdominal distension or peritonitis
Inconsolable crying (infants)
Neck stiffness + fever (meningitis)
Morning vomiting + headache (raised ICP)
These features suggest serious pathology such as:
Intestinal obstruction
Intussusception
Meningitis
Intracranial hypertension
Appendicitis
Step 3: Age-Based Differential Diagnosis
Age is one of the most powerful diagnostic clues.
1. Neonates (0–28 days)
Think danger first:
Intestinal obstruction (atresia, malrotation)
Hirschsprung disease
Sepsis
Inborn errors of metabolism
👉 Bilious vomiting = surgical emergency
2. Infants
Gastroesophageal reflux (common)
Pyloric stenosis → projectile vomiting
Intussusception
Food allergy
Infection
3. Children
Acute gastroenteritis (most common)
Appendicitis
UTI
Pneumonia (post-tussive vomiting)
Migraine
4. Adolescents
Pregnancy
Eating disorders
Drug/toxin ingestion
Diabetic ketoacidosis (DKA)
Intracranial causes
Step 4: Focused History
A good history often gives the diagnosis.
Key Questions
Onset
Sudden → infection, obstruction
Chronic → GERD, metabolic
Character of Vomit
Bilious → obstruction
Projectile → pyloric stenosis
Blood → gastritis, ulcer
Relation to Feeding
Immediately after feeds → reflux
Delayed → obstruction
Associated Symptoms
Fever → infection
Diarrhea → gastroenteritis
Headache → intracranial cause
Abdominal pain → surgical cause
Systemic Clues
Poor weight gain → chronic disease
Polyuria → DKA
Drug ingestion → toxins
Step 5: Physical Examination
General Examination
Hydration status:
Sunken eyes
Dry mucosa
Reduced urine output
Growth parameters (failure to thrive?)
Systemic Examination
Abdomen
Distension → obstruction
Tenderness → appendicitis
Mass → intussusception
CNS
Bulging fontanelle (infants)
Neck stiffness
Altered consciousness
Skin
Rash → infection/allergy
Petechiae → sepsis
Step 6: Investigations
👉 No “routine panel” exists — investigations should be targeted.
Basic Investigations
Serum electrolytes
Blood glucose
Blood gas (if severe)
When Indicated
Imaging
X-ray abdomen → obstruction
Ultrasound → intussusception
CT/MRI → CNS causes
Other tests
Urine analysis → UTI
LFT/RFT → systemic disease
Metabolic screening
Step 7: Management Approach
1. Treat the Cause (Definitive)
Surgery → obstruction
Antibiotics → infection
Insulin → DKA
2. Correct Dehydration (MOST IMPORTANT)
Mild–Moderate:
Oral Rehydration Therapy (ORT)
Severe:
IV fluids (bolus + maintenance)
3. Symptomatic Treatment
Ondansetron for persistent vomiting
NG decompression in obstruction
Electrolyte correction
4. Nutritional Support
Early feeding when tolerated
Continue breastfeeding
Step 8: Clinical Patterns to Recognize (Exam Gold)
Pattern
Likely Diagnosis
Projectile, non-bilious
Pyloric stenosis
Bilious vomiting
Intestinal obstruction
Vomiting + diarrhea
Gastroenteritis
Vomiting + headache (morning)
Raised ICP
Vomiting + abdominal pain → later
Appendicitis
Episodic vomiting, symptom-free intervals
Cyclic vomiting
Common Pitfalls
Ignoring bilious vomiting
Missing appendicitis early
Assuming all vomiting = gastroenteritis
Not checking hydration status
Over-ordering unnecessary tests
Clinical Algorithm (Simple Mental Model)
Is the child sick? (ABC + red flags)
What is the age?
What is the type of vomiting?
What are associated symptoms?
Targeted investigations
Treat dehydration + cause
Key Takeaways
Most pediatric vomiting is benign and self-limiting
But always rule out life-threatening causes first
Age + vomiting type + red flags = diagnosis
Hydration management saves lives
Conclusion
Vomiting in children is a diagnostic puzzle—but a structured approach simplifies it. The goal is not to memorize hundreds of causes, but to quickly identify danger, localize the system involved, and act appropriately.
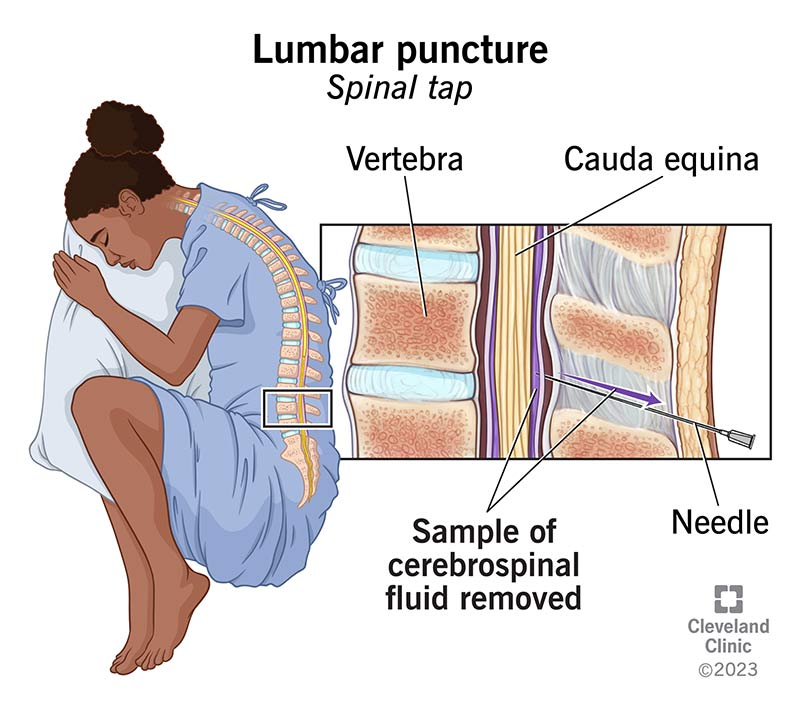
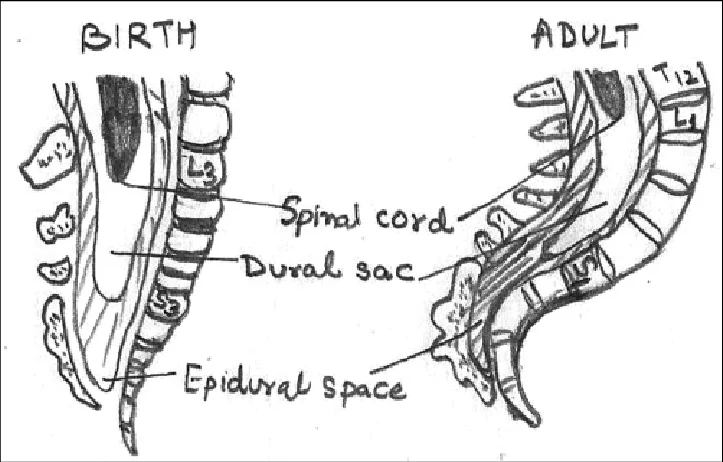
Lumbar puncture (LP) is a procedure in which a needle is inserted into the subarachnoid space of the lumbar spine to obtain cerebrospinal fluid (CSF) for diagnostic or therapeutic purposes.
Commonly done at L3–L4 or L4–L5 intervertebral space.
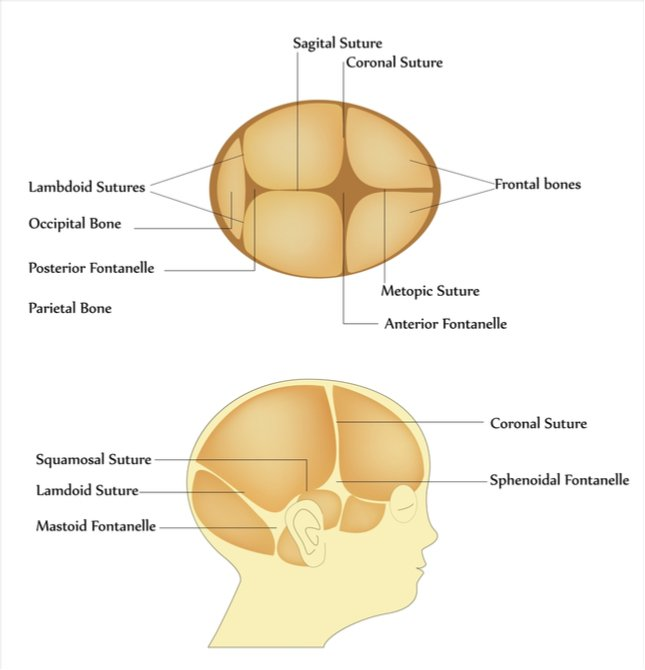
The anterior fontanelle is the largest fontanelle of the newborn skull and normally remains open during early infancy to allow brain growth and skull expansion.
1. Normal Anatomy and Physiology
The anterior fontanelle lies at the junction of:
Two frontal bones
Two parietal bones
Shape: Diamond-shaped
Average size at birth: 1–4 cm
Normal closure: 9–18 months
Functions:
Allows rapid brain growth
Facilitates molding during vaginal delivery
Serves as a clinical window for intracranial pressure assessment
When the Anterior Fontanelle Is Absent at Birth
A non-palpable or absent anterior fontanelle suggests premature fusion of cranial sutures or abnormal skull ossification.
This finding must always be evaluated carefully because it may indicate craniosynostosis or underlying pathology.
Causes of Absent Anterior Fontanelle
1. Craniosynostosis (Most Important Cause)
Premature fusion of one or more cranial sutures prevents normal skull expansion.
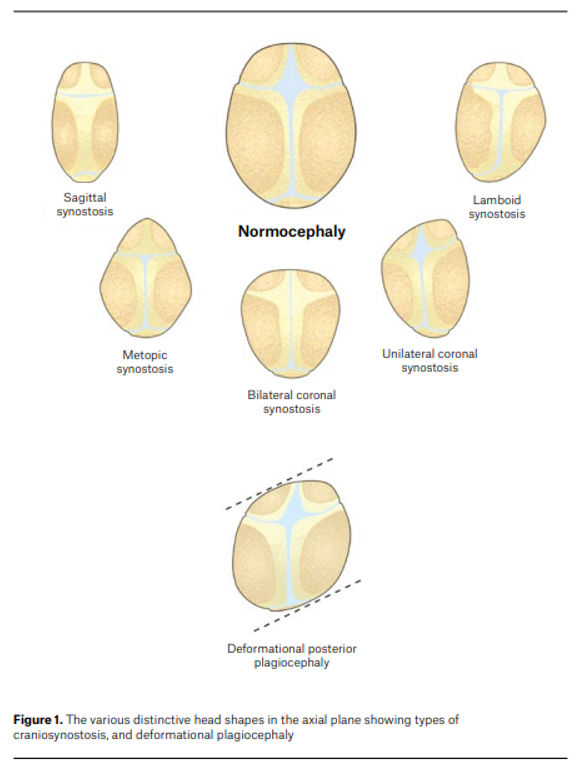
Types include:
Suture involved
Resulting head shape
Sagittal
Scaphocephaly (long narrow skull)
Coronal
Brachycephaly
Metopic
Trigonocephaly
Multiple sutures
Oxycephaly
Consequences:
Restricted skull growth
Raised intracranial pressure
Neurodevelopmental impairment if untreated
2. Hyperthyroidism (Congenital Thyrotoxicosis)
Seen in infants of mothers with Graves disease
Mechanism:
Increased thyroid hormone → accelerated bone maturation
Leads to early closure of sutures and fontanelles
Associated features:
Irritability
Tachycardia
Poor weight gain
Goiter
Exophthalmos (rare in neonates)
3. Microcephaly
Brain growth failure leads to small skull size, so sutures close early.
Common causes:
Intrauterine infections (TORCH)
Genetic syndromes
Severe hypoxic injury
Metabolic disorders
4. Skeletal Dysplasias
Some bone disorders cause abnormal skull ossification.
Examples:
Osteopetrosis
Thanatophoric dysplasia
5. Normal Variant
Rarely the fontanelle is very small or difficult to palpate, but sutures remain open and skull growth is normal.
Clinical Evaluation
1. History
Ask about:
Maternal history
Hyperthyroidism
Antithyroid drugs
TORCH infections
Perinatal history
Birth trauma
Neonatal illness
Family history
Craniosynostosis
Genetic syndromes
Developmental history
Feeding difficulty
Poor growth
Developmental delay
2. Physical Examination
Head Examination
Assess:
Feature
Significance
Head circumference
Detect microcephaly
Skull shape
Suggest specific craniosynostosis
Palpation of sutures
Check if fused or ridged
Remaining fontanelles
Posterior fontanelle status
Look for Associated Signs
Neurologic:
Irritability
Vomiting
Bulging veins
Systemic:
Signs of hyperthyroidism
Dysmorphic features
Investigations
1. Imaging
Skull X-ray
Shows fused sutures
Cranial ultrasound
If some fontanelle is open
CT scan with 3D reconstruction
Gold standard for diagnosing craniosynostosis
2. Laboratory Tests
If systemic cause suspected:
Test
Purpose
Thyroid function test
Detect neonatal thyrotoxicosis
TORCH screening
If infection suspected
Genetic testing
Syndromic craniosynostosis
Complications
If due to craniosynostosis:
Raised intracranial pressure
Visual impairment
Developmental delay
Cognitive impairment
Management
1. Craniosynostosis
Referral to pediatric neurosurgery
Treatment:
Surgical cranial vault remodeling
Usually performed within first year of life
2. Neonatal Hyperthyroidism
Treat underlying condition:
Antithyroid drugs
Beta-blockers
3. Microcephaly
Management depends on cause:
Developmental support
Treat underlying infection/metabolic disease
Clinical Pearls (High-Yield)
Anterior fontanelle absent at birth → think craniosynostosis first.
Always measure head circumference.
Check skull shape and sutures carefully.
3D CT scan confirms diagnosis.
Early surgical correction prevents intracranial hypertension and neurodevelopmental damage.